
一、细胞复苏
1.基本概念
细胞复苏是将冻存的细胞从低温状态(-80℃或液氮)快速解冻并恢复活性的过程。正确的复苏操作对细胞存活率和后续实验至关重要。
2.操作步骤
(1)复苏实验室环境的准备(细胞实验操作前均需确认环境)
确保超净台的清洁和消毒,使用75%酒精擦拭工作台面,并开启紫外灯消毒30分钟。
确保培养箱的温度为37℃,二氧化碳浓度稳定在5%左右,准备好预热的培养基和离心机,确保所有设备和试剂都处于最佳状态。
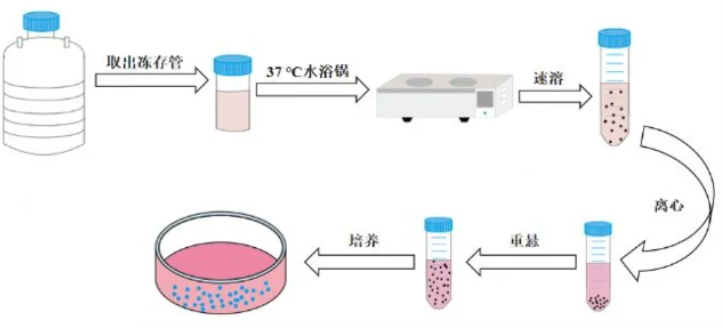
(2)细胞复苏(快融)
液氮罐中取出冻存管,立即放入37℃水浴锅中,轻柔摇晃冻存管,确保受热均匀。整个解冻过程应在1~2分钟内完成。如果解冻时间过长,细胞会受到热损伤,导致细胞死亡率增加。
解冻后的细胞需要离心,以去除冻存液中的DMS0。通常建议以2000rpm 离心3分钟。离心过程中,应注意避免剧烈震荡离心后,用移液器小心去除上清液,避免扰动细胞沉淀。
用预热的培养基重悬细胞沉淀后,转移细胞到培养瓶或培养皿中,并放入二氧化碳培养箱中培养。
在细胞贴壁后,需要更换新鲜培养基,以去除残留的DMS0和死细胞。
二、细胞冻存
1.基本概念
细胞冻存指通过将细胞置于低温环境中,减缓细胞代谢,从而实现细胞的长期保存
2.操作步骤
(1)细胞冻存液提前配置:
常用的细胞冻存液配比为培养基:血清:DMSO=7:2:1,适用于多数细胞系,能够保证细胞在冻存过程中有较高的存活率。
需要长时间保存或者具有特别珍贵性的细胞系调整为血清:DMSO的比例=9:1,这样可以更好地保护细胞,并确保其在冻存后依然保持较好的生物学特性。
(2)观察细胞状态
细胞密度在1-5x10~6个/ml之间。
细胞活力在90%以上(可以通过台盼蓝染色等方法检测)。
(3)冻存细胞
细胞满足要求后,弃培养基,加入1mI温浴PBS缓冲液清洗。
弃掉PBS,加入胰酶消化,消化时间根据细胞类型决定。
消化到时间后,加入二倍胰酶体积的培养基终止消化,用移液器将细胞全部吹下,转移至离心管内。
离心机常温2000rpm,离心5min。
弃上清,加入1mI配好的冻存液,移液器轻轻吹打混匀
冻存管标记好相应信息,包括细胞名称、代数、冻存时间操作者姓名等。将上述混悬液全部转移至冻存管内,拧紧冻存管管口,放入程序降温盒内。
将程序降温盒放入-80℃冰箱内,36h后可从程序降温盒内拿出细胞放在细胞冻存架上液氮保存。
3.注意事项
(1)细胞状态的评估:密度在1-5x10~6个/ml,活力90%以上。
(2)降温速率的控制:理想的降温速率是-1℃/min。缓慢降温可以使细胞有足够的时间通过渗透作用排出水分,减少冰晶的形成。如果降温过快,细胞内外的水分会迅速结冰,导致细胞膜和细胞器的损伤。因此,在冻存过程中,建议使用程序降温盒或梯度降温法,确保细胞在降温过程中受到的损伤最小。
(3)冻存环境的稳定性:液氮罐需要定期检查,确保液氮水平充足,避免液氮泄漏或干涸。建议每周检查液氮罐的液氮水平,并及时补充。同时,检查液氮罐的密封性,确保没有泄漏。液氮罐的温度应保持在-196℃左右,任何温度波动都可能导致细胞损伤。此外,液氮罐内的温度不均匀也可能导致细胞冻存效果不佳。因此,定期维护液氮罐,确保其稳定运行,是细胞冻存成功的关键
三、细胞传代
1.基本概念
细胞传代是体外细胞培养中,将生长到一定密度的细胞从原有培养容器转移到新容器中继续培养的操作过程。
2.细胞的传代数定义
(1)仅当完成“细胞转移-重新接种培养”核心流程,传代数才递增;若仅执行冻存,未开展复苏后接种,传代数保持不变。
(2)计数起点:原代细胞(从体内分离后首次培养)为P0;第一次传代后为P1,第二次传代后为P2,以此类推。若P3细胞经消化后分两瓶培养,两瓶均为P4;若P3细胞消化后冻存,3个月后复苏接种,复苏后细胞为P4。
3.准确记录细胞传代次数的意义
有限细胞系传代过多会衰老、功能异常;永生细胞系虽能无限传代,但长期传代可能积累变异,导致细胞特性改变(如增殖速率、分泌功能变化)。因此监测记录传代次数,能帮助我们把控细胞状态,保证实验结果的稳定性、可重复性,也能及时发现细胞是否发生转化等异常变化。
4.操作步骤
(1)显微镜下观察细胞密度长至90%左右,可以传代。
(2)弃细胞培养液沿侧面加入少量PBS,轻轻晃动使PBS充分接触细胞,冲洗后弃PBS。
(3)加入能盖住培养瓶底面细胞的胰酶,放入培养箱消化1分钟左右,显微镜观察状态,观察到细胞离散成单个圆形、细胞间出现间隙、呈沙状移动,并悬浮在培养液中即可。消化完成后(不同细胞消化时间有差异)立即加入完全培养基以终止消化。
(4)离心处理:将细胞悬液转移到离心管中,2000rpm离心3min弃去上清液。
(5)重悬细胞:加入适量的完全培养基重悬细胞,轻轻吹打混匀。
(6)接种新的培养瓶:根据需要的细胞密度,将细胞悬液分装到新的培养瓶中,补足培养基,轻轻混匀。
(7)培养箱培养:将接种好的培养瓶放入37℃,5%CO2的培养箱中培养,并标记细胞名称、传代数和传代日期。
四、细胞转染
1.基本概念
细胞转染是指将外源核酸(如DNA、RNA、SiRNA等)导入真核细胞内而获得新的表型的过程。转染后的细胞会产生重组蛋白,或者特异性的增强/抑制细胞中的基因表达。细胞转染是分子生物学、基因治疗、等领域的核心技术之一。细胞转染可以分为瞬时转染和稳定转染
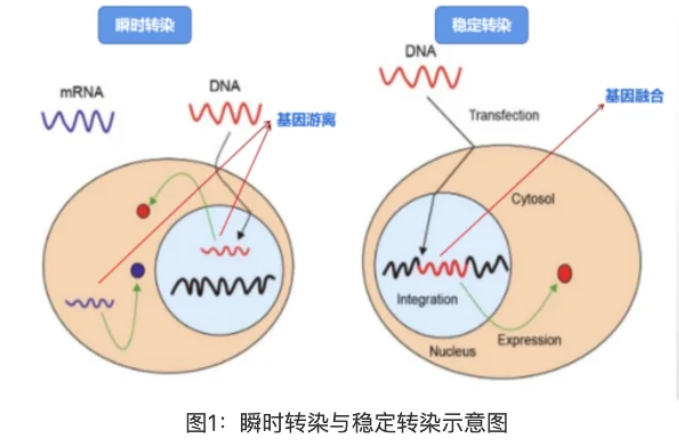
2.细胞转染类型
(1)瞬时转染:将外源核酸(如质粒DNA、mRNA等)导入真核细胞后,外源核酸不整合到宿主细胞的基因组中,仅在细胞内短暂表达的过程。其表达时间通常较短,一般持续数小时至数天,之后随细胞分裂或核酸降解而逐渐消失。
(2)稳定转染:外源核酸(主要DNA)导入真核细胞后,通过一定机制整合到宿主细胞的基因组中,并能随细胞分裂稳定传递给子代细胞,从而实现外源基因长期、持续表达的过程
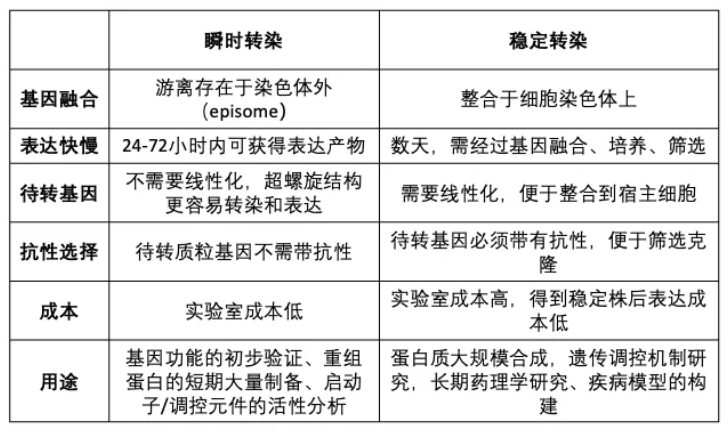
(3)瞬时转染和稳定转染的特点:
3.瞬时转染步骤
(1)实验前准备
试剂准备:阳离子脂质体、外源核酸、无血清培养基、含血清完全培养基、胰蛋白酶、PBS缓冲液(无钙镁)。
器材准备(需无菌处理):6孔板/12孔板(最常用,根据实验需求选择)培养皿、移液器(10μL/100μL/1000μL)、无菌吸头、离心管(1.5mL/15mL);超净工作台(无菌操作环境)、CO培养箱(37℃,5%CO2)、倒置显微镜(观察细胞状态)。
细胞准备(细胞状态良好):细胞处于对数生长期(活力>90%,无明显凋亡或污染);转染前1天,将对数生长期的细胞用胰酶消化,制成单细胞悬液;用含血清完全培养基调整细胞浓度,接种到6孔板中(每孔约2x105个细胞,体积2mL);放入CO2培养箱培养16~24小时,待转染时细胞汇合度达70%~80%(汇合度过低会导致转染效率低,过高会增加细胞毒性)转染前1小时,用无血清培养基清洗细胞1次,再加入1mL无血清培养基(减少血清对复合物的干扰)。
(2)转染(以6孔板转染质粒DNA为例)
制备脂质体 - 核酸复合物(关键步骤,需精准控时)
稀释核酸(DNA):取1.5m 无菌离心管,加入2μg质粒DNA(根据需求调整,6孔板每孔DNA用量通常为1~3μg),用无血清培养基(如0pti-MEM)补足至125μL轻轻混匀(避免剧烈震荡,防止DNA断裂)。
稀释脂质体:另取1.5mL 无菌离心管,加入4μL Lipofectamine 3000,用无血清培养基补足至125μL轻轻混匀后,室温静置5分钟(让脂质体充分分散,避免聚集)
形成复合物:将稀释好的脂质体溶液缓慢加入稀释后的DNA溶液中,轻轻吹打 3~5次(避免产生气泡);室温孵育15~20分钟(关键:孵育时间过短复合物未稳定,过长会导致复合物聚集,均降低转染效率)。
转染复合物加入细胞
用移液器将15~20分钟孵育后的复合物(共250μL)缓慢滴加到 6孔板的细胞孔中(每孔已含1mL无血清培养基)。
轻轻晃动培养板(左右/前后各3次),使复合物均匀分布在培养基中,避免局部浓度过高导致细胞毒性。
将培养板放入CO2培养箱(37℃,5%CO)中培养。
转染后换液(降低细胞毒性)
培养4~6小时后,吸弃含复合物的无血清培养基。
用PBS缓冲液清洗细胞1次。
加入2mL含血清的完全培养基(可含抗生素),继续在CO2培养箱中培养。
(3)转染效率检测
转染后24~48 小时进行检测,常用方法包括以下四种:
荧光观察(最直观):若质粒含荧光报告基因(如GFPRFP),用倒置荧光显微镜观察荧光细胞比例,荧光细胞占比越高,转染效率越高(贴壁细胞效率可达50%~90%)。
Western Blot:检测目的蛋白的表达量(需提取细胞总蛋白,适用于无荧光标签的质粒)。
qPCR:检测外源RNA的表达量(适用于siRNA/miRNA转染或检测目的基因的转录水平)。
流式细胞术:精准定量荧光细胞比例(适用于需要统计转染效率的实验)。
4.瞬时转染注意事项
(1)转染前的细胞状态和密度:对数生长期,汇合度70%~80%。避免支原体污染
(2)转染时的质粒纯度:带少量盐离子,蛋白、代谢物等污染的质粒会显著影响转染复合物的有效形成;含内毒素的质粒对细胞存在很大的毒性作用。转染过程中细胞通透性增加,抗生素可进入细胞降低其活性,导致转染效率降低。
(3)避免血清和抗生素干扰:制备复合物时必须用无血清培养基,转染前的细胞清洗和复合物孵育过程中,培养基不可含抗生素。
(4)DNA与转染试剂的比例:不同细胞的转染效率差异大,需提前优化DNA用量、脂质体用量、孵育时间等参数;首次实验可设置“空白对照”(仅细胞)、“脂质体对照”(仅加脂质体,检测毒性)、“阳性对照”(已知高表达的荧光质粒),排除非特异性干扰。
(5)操作细节控制:复合物孵育时间严格控制在15~20分钟(室温,不可超30分钟);加入复合物时动作轻柔,避免冲击细胞;转染后换液时间需根据细胞耐受度调整(若细胞出现明显皱缩,可提前至3小时换液)。
(6)转染后的筛选时间:转染后筛选不能太早或太晚,因为转染了外源基因的细胞代谢负荷大增殖较慢,太晚会被没有外源基因转入的细胞所淹没,最终导致筛选不出阳性克隆。
5.瞬时转染常见问题及解决方案
(1)转染效率极低
可能原因:细胞汇合度过低/过高;DNA纯度差(含内毒素);脂质体与DNA比例不当。
解决方案:调整细胞接种密度,确保转染时汇合度70%~80%;用无内毒素试剂盒提取DNA;按说明书优化比例(如1:1至3:1梯度测试)。
(2)细胞毒性高(大量细胞漂浮)
可能原因:脂质体用量过多;复合物孵育时间过长;转染后未及时换液。
解决方案:减少脂质体用量;缩短复合物孵育时间(≤20分钟);转染后4小时内换液。
(3)荧光细胞少但细胞状态正常
可能原因:质粒构建错误(如启动子失效);转染后培养时间不足。
解决方案:用阳性对照质粒验证(如PEGFP-N1);延长培养至48~72小时(部分细胞表达峰值较晚)。
(4)复合物聚集(培养基中有沉淀)
可能原因:脂质体或DNA浓度过高;混合时剧烈震荡。
解决方案:降低脂质体/DNA浓度,确保混合后体积足够;轻柔吹打混合,避免气泡。
细胞实验外包 想了解更多请关注:https://www.do-gene.cn/